|
Синдром Ангельмана (AS)Часть 2 (клиника и диагностика)В статье представлены основные клинические проявления синдрома Ангельмана (AS). Статья содержит современные данные об особенностях физического, интеллектуального, речевого и полового развития больных с AS...Резюме В статье представлены основные клинические проявления синдрома Ангельмана (AS). Статья содержит современные данные об особенностях физического, интеллектуального, речевого и полового развития больных с AS. Представлены данные о взаимосвязи клинических особенностей AS с характером генетических нарушений. В статье приведены клинические критерии диагностики синдрома Ангельмана с учетом частоты встречаемости основных клинических признаков. Авторами рассмотрено целевое назначение молекулярно-генетических анализов, используемых для верификации диагноза и определения генетического механизма AS. Статья содержит алгоритм лабораторно-диагностических действий проведения молекулярно-генетического обследования пациентов с AS. |
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ СИНДРОМА АНГЕЛЬМАНА Больные с синдромом Ангельмана (AS) рождаются практически здоровыми. Физическое развитие соответствует гестационному возрасту. Основные клинические проявления AS представлены ниже. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ AS Постоянные проявления: — умственная отсталость; — нарушение моторики; — задержка речевого развития; — особенности поведения (приступы немотивированного смеха, гиперактивность и дефицит внимания, счастливое выражение лица). Часто встречаемые признаки: — постнатальная микроцефалия с плоским затылком; — судороги; — затруднения при вскармливании, гастроэзофагеальный рефлюкс, запор; — нарушение сна; — постоянное слюнотечение; — пронации стопы. Редко встречаемые признаки: — сколиоз; — гипопигментация; — повышенная чувствительность к температуре окружающей среды; — нарушение развития длины тела в зависимости от генотипа. Малые аномалии развития: у больных с AS часто отмечаются черепно-лицевые аномалии. Практически у всех пациентов наблюдается постнатальная микроцефалия, у трети детей — с гипоплазией средней части лица, плоский затылок с «канавкой», глубоко посаженные глазные яблоки, широкий и часто постоянно открытый рот, заостренный подбородок. Физическое развитие: с течением возраста наблюдается замедление прироста окружности головы, и к двум годам жизни у 80 % пациентов диагностируется постнатальная микроцефалия. Психомоторное развитие: задержка психомоторного развития становится клинически значимой во второй половине первого года жизни. Примерно 10 % детей с AS не приобретают навыков самостоятельной ходьбы. Способность сидеть появляется после 12 месяцев жизни, ходить — после двухлетнего возраста, некоторые больные начинают ходить только к шести годам жизни. У детей отмечается своеобразная широкая походка. Больные широко расставляют ноги с повернутыми наружу стопами. Из-за резких, «рубленных» движений приподнятыми и согнутыми в локтевых суставах руками, которые помогают удерживать равновесие, походка напоминает походку робота. В раннем детском возрасте отмечаются гиперкинетические движения туловища и конечностей. Дрожание конечностей может наблюдаться уже в первые шесть месяцев жизни. Гиперкинезии, как правило, выраженные и резкие, которые мешают ходить и принимать пищу. Интеллектуальная недостаточность: при AS, как правило, наблюдается тяжелая умственная отсталость. У большинства больных когнитивное развитие практически не превосходит уровня, соответствующего 24–30-му месяцу жизни ребенка. Задержка речевого развития: выраженное отставание речевого развития является одним из ключевых признаков AS. У большинства больных отсутствует речь. Некоторые пациенты обладают очень бедным словарным запасом, и только отдельные больные могут изъясняться фразами. Больные, несмотря на задержку речевого развития, слушают и хорошо понимают обращенную к ним речь. Речевой дефицит дети c AS компенсируют невербальными средствами коммуникации. Судороги: судороги являются одним из наиболее часто встречаемых симптомов AS. Первые пароксизмы судорог могут инициироваться в различные возрастные периоды — от трехмесячного до 20-летнего возраста. В 75 % случаев первый приступ судорог отмечается до трехлетнего возраста. Судороги появляются практически у 80–95 % пациентов в течение первых трех лет жизни, на первом году жизни — только у 25 % больных. Часто судорожный синдром у больных с AS манифестирует с фебрильных судорог. Наиболее характерными проявлениями эписиндрома у детей раннего возраста являются миоклонические приступы (25 %), атонические припадки (23 %), генерализованные тонико-клонические судороги (21 %) и атипичные абсансы (12 %). На первом году жизни возможны такие проявления, как атипичные абсансы и эпилептический миоклонус. У больных с AS достаточно часто встречается эпилептический статус (около 50 %), который может продолжаться на протяжении нескольких дней, недель, а возможно и месяцев. Наиболее типичными изменениями ЭЭГ при AS являются: 1) пробеги генерализованной ритмичной высокоамплитудной (часто более 300 мкВ) дельта-активности, обычно с амплитудным преобладанием в лобных отделах, нередко в сочетании с эпилептиформной активностью; 2) высокоамплитудная активность тета-диапазона (200 мкВ и более) с частотой 4–6 Гц, локализованная в задних отделах или диффузно; 3) изменения в задних отделах — ритмичная высокоамплитудная эпилептиформная активность частотой 3–4 Гц в виде спайков, комплексов острая-медленная волна, локализованных преимущественно в затылочных областях. Эти изменения провоцируются закрыванием глаз и могут быть асимметричными. Нарушение сна: большинство детей с AS (80 %) отличаются низкой потребностью в сне. Продолжительность сна редко превышает 5–6 часов в сутки, однако в дневное время суток они испытывают чувство сонливости. Ночной сон беспокойный, часто прерываемый пробуждениями. При пробуждении больные проявляют высокую активность, настойчиво требуют к себе повышенного внимания, привлекая его самыми разнообразными способами. Нарушение ритма сон — бодрствование сопровождается снижением уровня мелатонина в сыворотке крови. Shu-qun Shi и соавт. показали, что именно нарушение импринтинга гена Ube3a лежит в основе нарушения циркадного ритма функционирования головного мозга. Мышечная дистония: Для детей с AS характерно сочетание гипотонии мышц туловища и гипертонии мышц конечностей в сочетании с гиперрефлексией сухожильных рефлексов. Затруднения при кормлении: затруднения при кормлении могут проявляться уже в первые месяцы жизни. При кормлении грудью у детей с AS отмечаются несогласованные движения языка, мышц губ и щек, участвующих в сосании. Перорально-моторные нарушения приводят к неэффективности кормления грудью. Стереотипии: у больных с AS наблюдаются необычные движения конечностями, которые включают в себя такие стереотипии, как хлопанье в ладоши, размахивание руками, подергивания конечностей. Атаксия: атаксия является практически постоянным признаком (88 %) AS. Счастливое выражение лица: своеобразное выражение лица у больных с AS постепенно приобретает черты, характерные для умиротворенного, счастливого человека. Часто (60 %) встречается немотивированный и неуместный смех. Гиперактивность и дефицит внимания: у большинства пациентов отмечается выраженная гиперактивность. Они находятся как бы в непрерывной деятельности, постоянно держат в руках игрушки или перемещают их в пространстве. Дети с AS с большим трудом концентрируют свое внимание, и если сосредотачивают его на определенном объекте, то на очень короткий промежуток времени. Особенности поведения: поведенческие особенности больных с AS характеризуются дружелюбностью, веселым нравом, легкостью инициации смеха, невнимательностью. Характерной особенностью поведения является влечение к воде, особенно текущей из крана, которое наблюдается у 75 % больных с AS. Повышенный риск возникновения расстройств аутистического спектра, как правило, связан с дупликациями материнской, а не отцовской копии хромосомы 15q11-q13; при этом предполагают, что гиперэкспрессия гена UBE3A в нейронах головного мозга является патогенетическим фактором, определяющим развитие аутизма. Механизм развития аутистических расстройств при дупликации региона q11-q13 копии отцовской хромосомы 15 остается пока не объясненным процессом. Практически все расстройства аутистического спектра в той или иной мере могут наблюдаться у детей с AS (95 %). К расстройствам аутистического спектра относятся разнообразные формы аномального поведения, нарушения социального и коммуникативного взаимодействия со строгим ограничением интересов и повторяющимися поведенческими актами. Дети с AS могут кусаться, щипаться, хватать за волосы. Это деструктивное поведение не предназначено для причинения вреда, а связано с легкой возбудимостью, стремлением привлечь внимание или недостаточным контролем над движениями. Половое развитие: половое развитие происходит в обычные возрастные периоды. Мужчины и женщины, больные AS, способны к продолжению рода. Другие проявления: для больных с AS характерны: гастроэзофагеальный рефлюкс (71 %), запоры (75 %), высунутый язык, постоянное слюнотечение (74 %), повышенная чувствительность к высокой температуре окружающей среды. Продолжительность жизни: продолжительность жизни у больных с AS на 10–15 лет меньше средней продолжительности жизни человека. Описаны пациенты с AS, прожившие более 70 лет. |
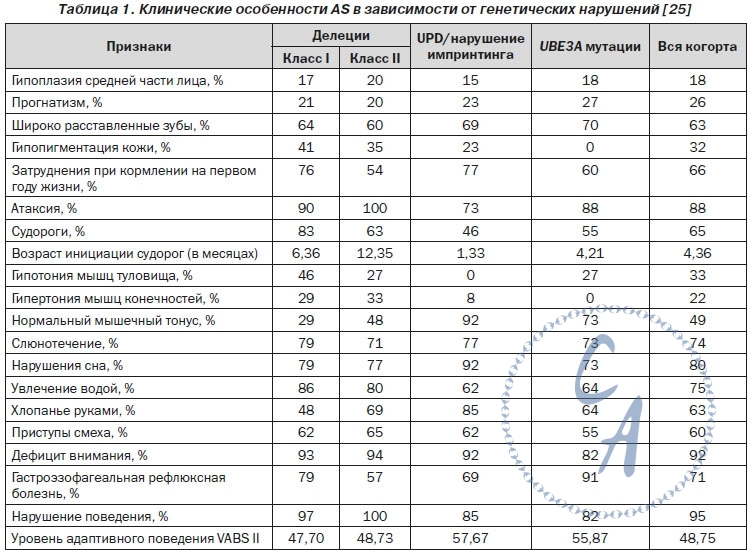
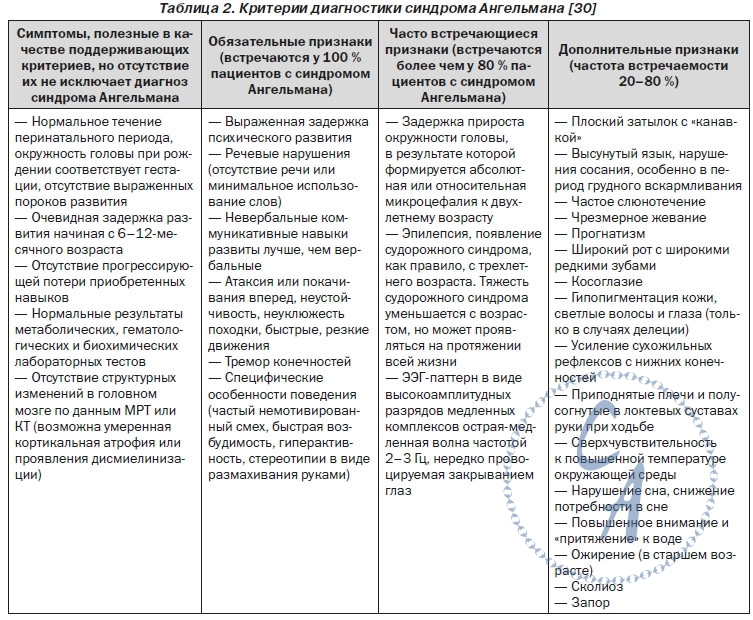
КОРРЕЛЯЦИЯ ФЕНО- И ГЕНОТИПА Некоторые клинические проявления AS коррелируют с характером нарушений генотипа (табл. 1) Большие делеции (от 5 до 7 мб) сопровождаются тяжелым проявлением заболевания в сочетании с недостаточной прибавкой массы тела. Больные с делецией I класса (от ВР1 до BP3) отличаются выраженной задержкой речевого развития, значительными проявлениями расстройств аутистического спектра и резистентностью к терапии в отличие от лиц с делецией II класса (от ВР2 до BP3). У пациентов с отцовской дисомией AS протекает более легко — со значительно меньшей вероятностью развития микроцефалии, эпилепсии, атаксии. У больных с делецией гена OCA2 (oculocutaneous albinism II) наблюдаются фенотипические признаки нарушения меланинового обмена — гипопигментация кожи, волос, глазных радужек. КРИТЕРИИ ДИАГНОСТИКИ Клинические критерии диагностики AS были разработаны специалистами Scientific Advisory Committee of the US Angelman Syndrome Foundation (табл. 2) |
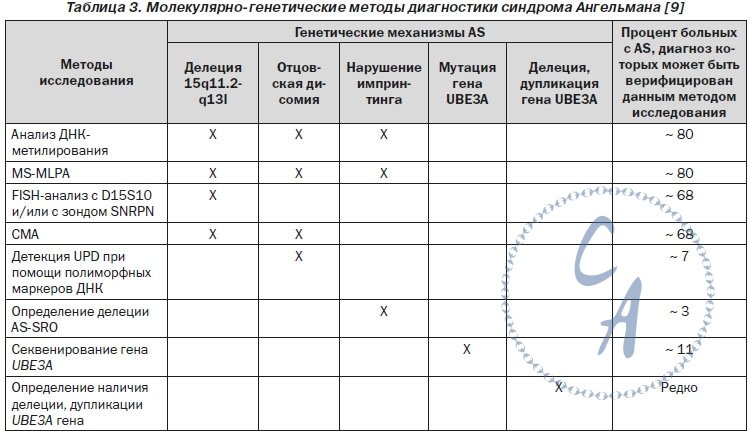
МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКАЯ ДИАГНОСТИКА Основными методами верификации диагноза AS являются молекулярно-генетические методы исследования — анализ метилирования ДНК (метил-специфическая ПЦР (MS-PCR), метил-специфическая мультиплексная лиганд-зависимая амплификация (MS-MLPA), флуоресцентная гибридизация in situ (FISH-анализ), хромосомный анализ микрочипов (CMA), детекция отцовской изодисомии при помощи полиморфных маркеров ДНК, микрочипированный ген-таргетный анализ, анализ последовательности гена UBE3A и другие. Анализ метилирования ДНК и анализ последовательности гена UBE3A позволяют обосновать диагноз приблизительно у 90 % пациентов. Молекулярно-генетические методы, используемые для верификации диагноза и определения генетического механизма AS, представлены в табл. 3. |
|
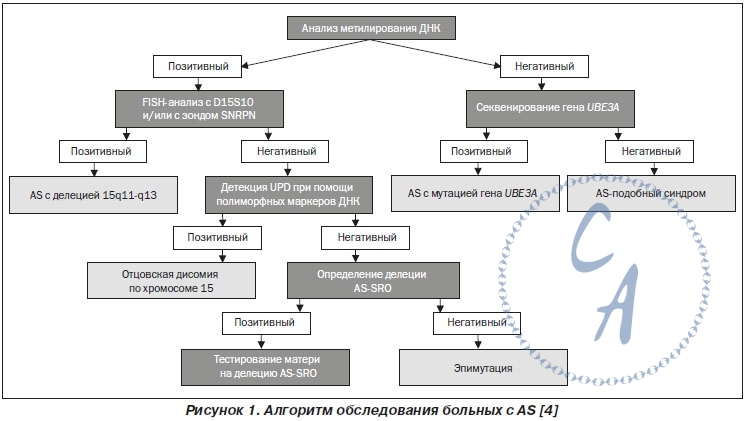
Наиболее чувствительным методом для диагностики AS является исследование метилирования ДНК в области 15q11-q13, в частности, дифференциально метилированного экзона-1/промоторной области гена SNURF-SNRPN. У здоровых людей метилирован материнский аллель и не метилирован отцовский аллель. У пациентов с AS, заболевание которых обусловлено de novo делецией в области 15q11-q13 материнского аллеля, отцовской дисомией 15 или дефектом импринтинга, находят только неметилированный отцовский аллель. Пациенты с мутациями гена UBE3A характеризуются нормальной структурой метилирования. Метод MS-MLPA может не только обнаруживать дефект метилирования, но и различать делеции 15q11-q13 и импринтингового центра. Но MS-MLPA не идентифицирует отцовскую дисомию по хромосоме 15 и нарушения импринтинга, которые не обусловлены делецией, в связи с чем дальнейшее тестирование требует микросателлитного анализа ДНК. При проведении исследования методом MLPA рекомендуют использовать SALSA MLPA KIT ME028-B1 (MRC-Holland) или диагностические комплекты других производителей, которые содержат зонды, специфичные для региона 15q11-13. При проведении молекулярно-генетического обследования пациентов с AS рекомендуется использовать последовательность лабораторно-диагностических действий, представленную на рис. 1. |
Пренатальная диагностикаПроведение пренатальной диагностики рекомендуется в случаях молекулярно-генетического подтверждения AS у предыдущего ребенка в семье. Особенное значение пренатальная диагностика имеет в случаях с высоким риском рождения ребенка с AS — при отцовской дисомии по хромосоме 15 с транслокацией, несбалансированной транслокацией хромосомы 15, делеции импринтингового центра. Анализ ДНК проводят в клетках биоптата хориона или амниоцитах. Некоторые клинические лаборатории из-за относительного гипометилирования плацентарных клеток предпочитают использовать амниоциты для проведения анализа метилирования ДНК в процессе пренатального тестирования. Основными методами молекулярно-генетического исследования при проведении пренатальной диагностики AS являются FISH, CMA, анализ метилирования ДНК, MS-MLPA и методы обнаружения однонуклеотидных полиморфизмов.
http://www.mif-ua.com/archive/article/41852
#Семьям особенных детей нужна Ваша помощь#Благотворительный Фонд «Синдром Ангела» |
|
|
|
|
|
|
|
 Помочьдетям.
Помочьдетям.