|
Синдром Ангельмана (AS)Часть 1 (этиология и патогенез). В статье приведены современные представления о механизмах генетических изменений при синдроме Ангельмана... Резюме. В статье приведены современные представления о механизмах генетических изменений при синдроме Ангельмана. Статья содержит данные о частоте встречаемости и риске наследования различных генетических дефектов у больных с синдромом Ангельмана. Представлены патогенетические основы развития основных клинических проявлений синдрома Ангельмана: судорожного синдрома, когнитивного дефицита, расстройств поведения, нарушения баланса времени бодрствования и сна, трофических нарушений. Показано, что изменения экспрессии гена Arc не только ассоциированы с синдромом Ангельмана, но и сопровождают некоторые другие заболевания. |
Определение. Синдром Ангельмана (синдром «счастливой куклы» или «счастливой марионетки», Angelman syndrome — AS; OMIM: 105830) — генетическое заболевание, в основе которого лежат морфологические или функциональные нарушения локуса q11-q13 копии материнской хромосомы 15, сопровождающиеся потерей экспрессии гена UBE3A в нейронах головного мозга. Синдром Ангельмана клинически проявляется интеллектуальным дефицитом, задержкой речевого развития, расстройствами развития нервной системы, судорогами и специфическим поведенческим профилем. Впервые данное заболевание описал английский педиатр Harry Angelman в 1965 году, когда он обобщил фенотипы трех умственно отсталых детей, которые были охарактеризованы как puppet children — «дети-куклы» из-за необычной позиции рук и свое образных дерганных и отрывистых движений конечностей (с изоляцией отдельных фаз движений и фиксацией промежуточных положений конечностей). |
|
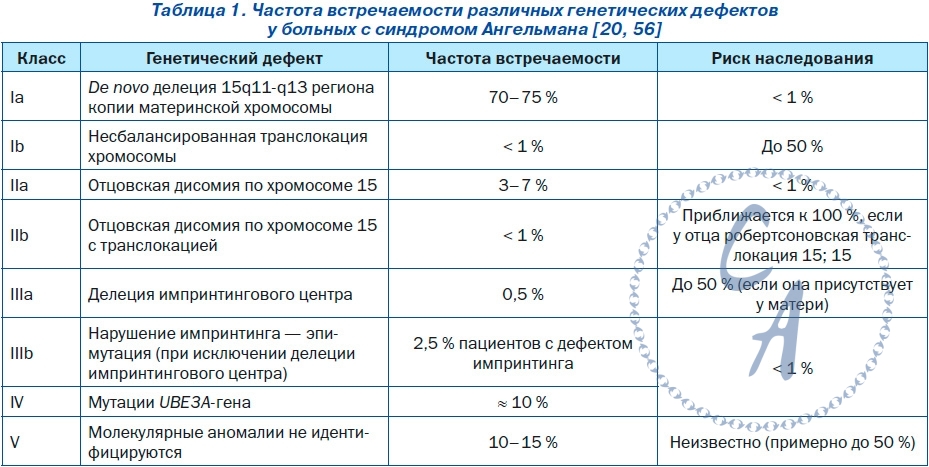
Эпидемиология. Уровень распространенности AS колеблется в пределах 1 : 10 000 и 1 : 20 000 населения. Этиология. Различают 4 генетических варианта AS (табл. 1): 1) de novo делеция в локусе 15q11-q13; 2) отцовская дисомия по хромосоме 15; 3) дефект центра импринтинга;4)мутация материнской копии гена убиквитинлигазы UBE3A Делеции. Различают два класса делеций области q11-q13 хромосомы 15: I класс — делеция от ВР1 до BP3 II класс — делеция от BP2 до BP3. Делеция I класса имеет геномную длину 6,58 Мб, и ее доля в структуре AS-ассоциированных делеций составляет около 40 %, делеция II класса (5,33 Мб) встречается при-близительно в 50 % случаев. Чаще всего микроделеции происходят в области геномного кластера, которая располагается между ВР1 и BP2, BP3 и BP4 Отцовская однородительская дисомия по хромосоме 15. В структуре генетических причин AS 3–4 % приходится на отцовскую однородительскую дисомию по хромосоме 15 (uniparental рaternal disomy (UPD) 15), которая сопровождается невыраженными фенотипическими проявлениями с низкой частотой встречаемости судорожных пароксизмов и хорошим физическим развитием больных. Основным механизмом формирования отцовской UPD по хромосоме 15 является нарушение соматической сегрегации хромосом. |
Нарущение импиринга. Нарушение импринтинга кластера PWS, которое приводит к развитию AS, обусловлено или делецией дифференциально метилированной области импринтингового центра AS-SRO, или нарушением дифференциальности ее метилирования. Аномальное метилирование AS-SRO отмечается у 2–4 % больных с AS. Примерно у 10–15 % больных, развитие AS у которых обусловлено нарушением импринтинга, отмечается делеция области AS-SRO на копии материнской хромосомы |
|
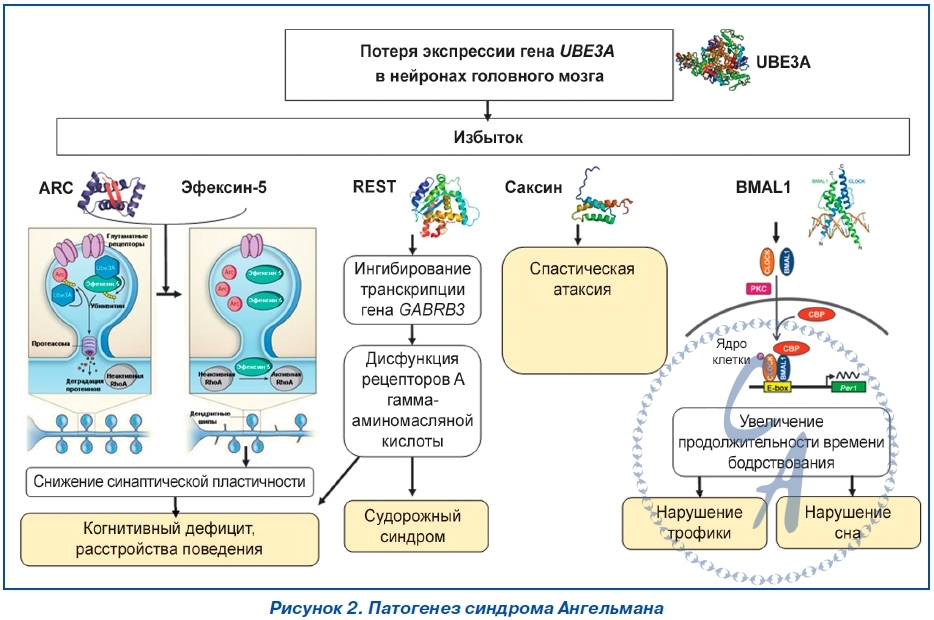
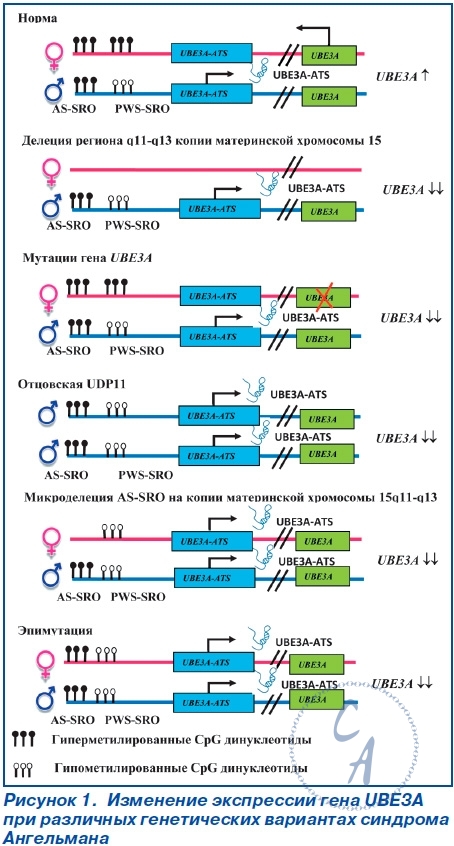
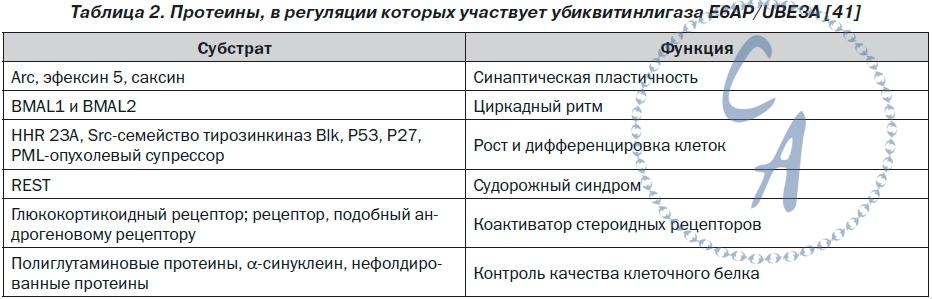
Мутации гена UBE3A.Большинство мутаций гена UBE3A у лиц с AS сопровождается продукцией функционально неактивного протеина, лишенного домена HECT. Мутации гена UBE3A представлены инсерциями, микроделециями, миссенс-, нонсенс- и сплайсинговыми мутациями. В настоящее время идентифицировано более 60 мутаций гена UBE3A, из которых 60–70 % представляют небольшие делеции и дупликации. Приблизительно 25 % структурных нарушений гена UBE3A — это миссенс- и нонсенс-мутации. Патогенез.Ключевой причиной развития AS определен дефицит экспрессии гена UBE3A на копии материнской хромосомы 15 в нейронах головного мозга (рис. 1). Ген UBE3A не является дифференцированно метилированным; импринтированность его экспрессии косвенно регулируется длинной антисмысловой некодирующей РНК (UBE3A-ATS). Транскрипт UBE3A-ATS является большой (> 600 кб) РНК, синтез которой инициируется в области управления импринтинга PWS-SRO. Экспрессия и/или процессинг UBE3A-АТС в ткани головного мозга и других тканях организма существенно отличается друг от друга. Транскрипт UBE3A-АТС обнаруживается во всех тканях, но только в нейронах распространяется в дистальном направлении и перекрывает ген UBE3A. Антисмысловая РНК UBE3A-АТС блокирует экспрессию гена UBE3A cis-зависимым образом. Изменения в уровне метилирования ДНК и ацетилирования гистоновых белков PWS-SRO могут приводить к активности экспрессии UBE3A-АТС. Механизм, посредством которого транскрипта UBE3A-АТС блокирует транскрипцию гена UBE3A, остается неизвестным. Предполагают, что транскрипта UBE3A-АТС может индуцировать гистонопосредованную репрессию транскрипции. Ген UBE3A (120 кб) кодирует протеин убиквитинлигазу Е3A (ubiquitin protein ligase E3A) — E6AP/UBE3A, основной функцией которого является участие в процессе деградации протеинов через убиквитинпротеасомный путь. Первоначально протеин E6AP/UBE3A был определен как протеин, взаимодействующий с E6-вирусным белком, кодируемый геномом вируса папилломы человека 16 (HPV16). Большинство (80–90 %) внутриклеточных белков расщепляются при помощи 26S-протеасомы. Протеасома отбирает для деградации только те протеины, которые конъюгированы с полиубиквитином. Убиквитинирование протеинов — ковалентное присоединение убиквитина к белковому субстрату — выполняют специфические убиквитинлигазы. Протеин E6AP/UBE3A является пред-ставителем семейства протеинов домена HECT. Его С-терминальный каталитический домен HECT характеризуется наличием сайта активного цистеинового остатка, образующего тиоэфирную связь с убиквитином для последующей его передачи специфическим белковым субстратам. Спектр протеинов, с которыми взаимодействует E6AP/UBE3A, определяет его физиологические функции (табл. 2) Протеин E6AP/UBE3A локализуется в ядре клеток, дендритных шипах (spine), пресинаптических и постсинаптических отделах синапсов нейронов. Снижение функциональной активности протеина UBE3A за счет дефицита его представительства или наличия неактивных форм из-за нарушения убиквитинирования приводит к снижению уровня деградации и избыточному накоплению его целевых субстратов. Так, дефицит протеина E6AP/UBE3A или продукция его мутантных форм, характеризуемых отсутствием домена HECT, приводит к нарушению деградации целевых для E6AP/UBE3A протеинов — цепей ионотропных глутаматных рецепторов, цито-скелет-ассоциированного протеина (cytoskeleton-associated protein — ARC), эфексина-5 (ephexin-5), саксина (sacsin). Необходимо отметить, что убиквитинлигаза E6AP/UBE3A не только способствует деградации протеина ARC, но и подавляет эстрадиол-индуцированную транскрипцию его гена ARC. Michel Baudry и соавторы считают, что дефицит активности протеина E6AP/UBE3A приводит к избытку представительства протеина ARC, ведущего к изменению организации цитоскелета дендритных шипов нейронов. Дендритные шипы представляют собой грибоподобные выпячивания длиной около одного микрона мембраны дендритных отростков. Более 90 % синапсов образуется на дендритных шипах. Большинство постсинаптических мишеней глутаматергических аксонов в головном мозге располагаются на дендритных шипах. Морфологические и функциональные изменения дендритных шипов могут лежать в основе развития наркомании, некоторых психоневрологических заболеваний, патологии процессов обучения и памяти. Так, установлено, что синдром фрагильной хромосомы Х и синдром Ретта сопровождаются развитием аномально длинных дендритных шипов и увеличением плотности их расположения на дендритных отростках клеток различных нейронных популяций. Протеин E6AP/UBE3A хотя и не регулирует нейрогенез как таковой, но локально участвует в формировании дендритных шипов и развитии синаптической пластичности. Дефицит протеина E6AP/UBE3A сопровождается формированием дендритных шипов преимущественно с короткими «шеями» и относительно низкой плотностью их распределения на дендритных отростках клеток Пуркинье мозжечка, пирамидных нейронов гиппокампа и коры головного мозга. Известно, что передача сигнала в глутаматергических синапсах осуществляется рецепторно-трансмиттерными системами, одним из центральных компонентов которых являются ионотропные глутаматные рецепторы N-метил D-аспартата (glu-tamate receptor, ionotropic, N-methyl D-aspartate (NMDA) — GRIN), α-амино-3-гидрокси-5-метил- 4-изоксазолпропионовой кислоты (α-amino-3-hydroxy-5-methyl-4-isoxazole propionate acid (AMPA) receptors/glutamate receptor, ionotropic — AMPAR/GRIA) и каината. В функционировании рецепторно-трансмиттерных систем также принимают участие Са2+/кальмодулинзависимая протеинкиназа II (calcium/calmodulin-dependent protein kinase II — CAMK2/CaM-KII), discs, большой гомолог-4 (discs, large homolog-4 — DLG4/PSD-95) и протеин-1, активирующий Ras-GTPазу (synaptic Ras GTPase activating protein-1 — SYNGAP-1) постсинаптической зоны. Ионотропные глутаматные рецепторы NMDA регулируют транспорт ионов Na+, K+ и Ca2+, а рецепторы AMPA — транспорт только ионов Na+ и K+ и передают быстрые возбуждающие сигналы. Количество GRIA на постсинаптической мембране зависит от активности протеина ARC и эфексина-5, которые определяют скорость интернализации этих рецепторов. Недостаточность активности протеина E6AP/UBE3A обусловливает снижение протеасомной деградации протеинов ARC, эфексина-5 и избыточное их накопление в дендритных шипах, что приводит к снижению представительства AMPAR/GRIA и, как следствие, к нарушению синаптической пластичности — способности нейронов изменять и адаптировать синаптическую активность в зависимости от внешних воздействий. В норме адекватное содержание протеинов ARC и эфексина-5 обеспечивает равновесное состояние процессов синтеза и деградации AMPAR/GRIA, поддерживая необходи-мое представительство рецепторов AMPAR/GRIA в постсинаптической плотности. Рецепторы AMPAR/GRIA являются важнейшими молекулярными компонентами механизмов синаптической пластичности. Рецепторы AMPAR/GRIA представляют тетрамерные лиганд-зависимые ионные каналы, в формировании которых участвуют че-тыре рецепторных субъединицы (GluR1–GluR4). Большинство AMPAR/GRIA-нейронов головного мозга являются гетеротетрамерами, состоящими из GluR1- и GluR2-димеров. Согласно модели синаптического скейлинга, изменение активности постсинаптического нейрона обусловлено модификацией синаптических входов. Физиологическая сущность синаптического скейлинга заключается в том, что сила синаптической передачи увеличивается при снижении и после полного подавления нейрональной активности. Изменения синаптического входа могут влиять на интегративные свойства синапсов и потенциально модулировать синаптическую пластичность. Синаптический скейлинг в постсинаптическом нейроне сопровождается перестройкой передачи импульса, в частности изменением амплитуды миниатюрных возбуждающих постсинаптических токов, опосредованных AMPAR/GRIA. Эти изменения часто сопряжены с изменением состава и представительства синап-тических AMPAR/GRIA. Рецепторы AMPAR/GRIA играют важнейшую роль в развитии долговременной синаптической потенциации (long-term potentiation — LTP), которая характеризуется сохранением активности синапса на протяжении нескольких часов или суток после его кратковременной активации, и долговременной синаптической депрессии (long-term depression — LTD), проявляющейся снижением синаптической передачи после периода активности синапса. Различают раннюю и позднюю фазы LTP. Ранняя фаза LTP, как правило, длится 1–2 часа после индукции LTP и сопровождается посттрансляционными модификациями уже существующих протеинов и отсутствием синтеза новых протеинов. Ранняя фаза LTP обусловлена тем, что после связывания с глутаматом, который высвободился из пресинаптического нейрона, открываются каналы AMPAR/GRIA, пропускающие ионы Na+ внутрь клетки, что приводит к снижению мембранного потенциала. Снижение мембранного потенциала способствует активации рецепторов NMDA за счет высвобождения ионов магния. Активацию рецепторов NMDA обусловливает приток Ca2+ в клетку. Повышение уровня внутриклеточного содержания ионов Ca2+ изменяет активность множества сигнальных путей, функционирующих в постсинаптическом пространстве. Повышение внутриклеточной концентрации ионов Ca2+ сопровождается активацией протеинкиназы CaM-KII, экстрацеллюлярной сигнал-регулируемой протеинкиназы (extracellular signal-regulated protein kinase — ERK), протеинкиназы A (protein kinases A — PKA) и протеинкиназы С (protein kinases С — РКС). Данные сигнальные пути участвуют в регуляции эндосомальной интернализации рецепторов AMPAR/GRIA и модуляции динамики актинового цитоскелета в дендритных шипах. Их активация приводит к увеличению представления рецепторов AMPAR/GRIA на постсинаптической мембране и транзиторному расширению поверхности дендритных шипов. Для формирования стабильной поздней LTP, которая длится многие часы или даже дни, требуются экспрессия новых генов и синтез протеинов. Первый период синтеза протеинов происходит в течение первых двух часов после индукции LTP. Поздняя LTP связана со стабильной реконструкцией постсинаптической плотности (мембран-ассоциированного большого мультибелкового комплекса), увеличения уже существующих дендритных шипов, а также de novo формирования синапсов. |
|
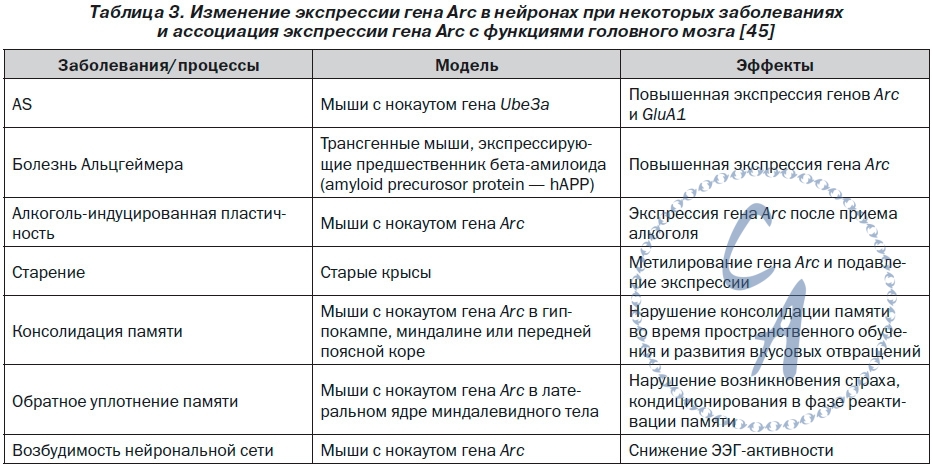
В основе LTD лежит уменьшение молекулярного представительства рецепторов AMPAR/GRIA в постсинаптических участках дендритных шипов. Долговременная синаптическая депрессия может быть вызвана активацией метаботропных глутаматных рецепторов (mGluR) и NMDAR. Выраженность LTD зависит от активации фосфатаз — серинтреониновой фосфатазы (serine-threonine protein phosphatase — PP1), кальциневрина — и сопровождается удалением AMPAR/GRIA с постсинаптической мембраны. Долговременные LTP и LTD ассоциированы с механизмами памяти и забывания. Избыточное накопление протеинов Arc и эфексина-5 обусловливает ускорение интернализации рецепторов AMPAR/GRIA, которое сопровождается уменьшением их представительства на постсинаптических участках, что приводит к существенному дефициту LTP и усилению LTD. В эксперименте показано, что AS сопровождается усилением экспрессии гена Arc. Изменения экс-прессии гена Arc ассоциированы с проявлениями и некоторых других заболеваний (табл. 3) Протеин Arc через взаимодействие с другими белками участвует в регуляции синаптической пластичности. В частности, протеин Arc образует комплекс с эндорфином и динамином, который способствует увеличению скорости эндоцитоза ионотропных глутаматных рецепторов AMPAR/GRIA. Комплекс протеина Arc с динамином расщепляет рецептор протеина Notch, активируя Notch-ассоциированные сигнальные пути, которые также участвуют в поддержании синаптической пластичности. Ключевое значение протеина Arc в поздней фазе LTP и LTD подчеркивает его функциональную роль как в процессе обучения, так и в формировании долгосрочной памяти. Показано, что высокий уровень содержания протеина Arc в области CA1 гиппокампа сопровождается потерей долгосрочной памяти у взрослых особей крыс. РНК Arc появляется в ядре нейрона через несколько минут после нейронной активации клетки и удерживается в цитоплазме около 30 минут, высокая концентрация и длительное ее сохранение, вероятно, поддерживают неадекватное возбуждение нейрона. Экспериментальные крысы с высоким уровнем экспрессии гена Arc в нейронах гиппокампа отличаются более медленным темпом обучения. Другим механизмом, который обусловливает дефицит LTP при AS, является аномальное фосфорилирование Thr286 и Thr305 молекулы CAMK2, которое ингибирует ее активность. Низкая активность CAMK2 приводит к недостаточному фосфорилированию субъединиц AMPAR, тем самым уменьшая проводимость ионных каналов. Са2+/кальмодулин-зависимая протеинкиназа II имеет решающее значение в индукции LTP, определяющей долгосрочную память. Установлено, что у мышей со сниженной экспрессией гена UBE3A в нейронах Пуркинье наблюдается повышение активности комплекса 1 серин-треониновой киназы mTOR (mammalian target of rapamycin complex 1 — mTOR C1) и уровня фосфорилирования его субстрата — S6 киназы-1 (S6K-1) в сочетании со снижением активности mTORC2 и уровня фосфорилирования его субстратов — AKT и N-Myc. Известно, что серин-треониновая ки-наза mTOR может взаимодействовать с протеинами Raptor (regulatory associated protein of mTOR) и Rictor (rapamycin-insensitive companion of mTOR). Взаимо-действие mTOR с протеином Raptor приводит к образованию чувствительного к действию рапамицина комплекса mTORC1, а с не чувствительным к действию рапамицина протеином Rictor — комплекса mTORC2. Комплекс mTORC1 также содержит про-лин-богатый Akt/PKB-субстрат 40 кДа (proline-rich Akt/PKB-substrate 40 kD — PRAS40), а комплекс mTORC2 — протеин, взаимодействующий с SAPK1 (SAPK interacting protein-1 — SIN1), и белок Protor (protein observed with rictor). Комплекс mTORC1, активируя киназу S6K (ribosomal protein S6 kinase) и репрессируя протеин, связывающий фактор инициации трансляции eIF4E, 4E-BP (eukaryotic translation initiation factor 4E binding protein 1), участвует в регуляции трансляции. Таким образом, комплекс mTORC1 является ключевым регулятором синтеза протеинов и роста клеток, а также необходим для формирования долговременной памяти. Комплекс mTORC2 возбуждает сигнальные пути, которые участвуют в поддержании цитоскелета клетки. Основным физиологическим эффектом активации mTORC1 в постсинаптической плотности является повышение высвобождения нейромедиаторов из пресинаптических нейронов. Так, установлено, что гиперактивация mTORC1 сопровождается общим увеличением пресинаптического высвобождения как возбуждающего, так и тормозящего нейромедиатора — глутамата и гамма-аминомасляной кислоты (ГАМК) соответственно. Следует отметить, что гиперактивация mTORC1 не приводит к изменениям LTP или LTD. Однако высокий уровень mTORC1 коррелирует с инициацией судорог, а ингибиторы mTORC1 обладают противоэпилептической активностью. Комплекс mTORC2 необходим для стабилизации ряда таких AGC киназ, как AKT, PKC и SGK1, в связи с чем отсутствие mTORC2 (нокаут гена Rictor) не совместимо с жизнью. Wei Huang и соавторы показали, что специфическое удаление mTORC2 в возбуждающих нейронах, локализующихся в лимбической и корковых областях, исключает возможность развития поздней фазы LTP и сопровождается ухудшением эффективности обучения, активности долгосрочной памяти. Делеция гена Rictor в нейронах переднего мозга мышей сопровождается значительным снижением активности mTORC2 в сочетании с ингибированием полимеризации F-актина. Авторы считают, что mTORC2 является потенциальной мишенью для возможных терапевтических средств, которые могли бы быть использованы для лечения когнитивных расстройств. Таким образом, когнитивный дефицит при AS обусловлен тем, что отсутствие E6AP/UBE3A при-водит к невозможности модулировать активность синапсов или перелокализовывать синапсы в соответствии с требованиями деятельности. Дефицит убиквитинлигазы E6AP/UBE3A сопровождается накоплением саксина. Показано, что мутация c.11,104A > G гена SACS, которая приводит к образованию аномального протеина саксина, лишенного сайтов связывания с убиквитинлигазой E6AP/UBE3A, фенотипически проявляется аутосомно-рецессивной спастической атаксией Шарлевуа — Сагене (autosomal recessive spastic ataxia of Charlevoix — Saguenay — ARSACS). Учитывая, что наиболее часто у больных с AS делецированная часть области q11-q13 хромосомы 15 содержит кластер генов трех субъединиц⍺ 5, β 3 и λ 3 рецептора A гамма-аминомасляной кислоты (gamma-aminobutyric acid (GABA) A receptor — GABR), развитие судорожного синдрома у данных пациентов связывают с дисфункцией GABR. ГАМК образуется при декарбоксилировании глутаминовой кислоты глутаматдекарбоксилазой. ГАМК активизирует два основных типа рецепторов: GABR, которые функционируют как хлоридные каналы, и чувствительные к баклофену метаботропные рецепторы GABRB. Рецепторы GABR представляют собой пентамерные каналы, состоя-щие из различных комбинаций нескольких субъе-диниц: ⍺ (1–6), β(1–3), λ(1–3), δ,ε,θ ,ω. Обязательными компонентами GABR являются четыре субъединицы — две ⍺- и две β-цепи, пятая субъединица может принадлежать к любому другому классу субъединиц. Сочетание субъединиц определяет фармакологические и фармакокинетические свойства, в том числе профиль агонистов, аффинитет к аллостерическим модуляторам или антагонистам и субклеточную локализацию. Сайт связывания GABR формируют⍺- и β-субъединицы. Активация GABR оказывает ингибирующее действие на активность нейронов. В упрощенном виде действие ГАМК может быть представлено в виде следующей последовательности событий: возбуждение GABR - импорт ионов Cl- в нейрон - гиперполяризация клетки - ингибирование нейропередачи. Дисфункция GABR приводит к нарушению притока в клетку ионов Cl-, что обусловливает неконтролированное повышение активности нейронов и развитие судорожного синдрома. Наиболее существенным фактором, определяющим развитие судорог, является делеция гена GABRB3 или нарушение функционирования субъединицы β3 GABR.
|
|
Дефицит протеина E6AP/UBE3A также приводит к увеличению репрессорного фактора REST (RE1-silencing transcription factor). Протеин REST первоначально был идентифицирован как фактор транскрипции, который подавляет гены, содержащие репрессорный элемент-1/нейрон, ограничивающий сайленсорный элемент (RE1/NRSE) cis-регуляторной последовательности ДНК-промоторной области. В связи с этим повышен-ная концентрация репрессорного фактора REST в нейронах приводит к подавлению транскрипции гена GABRB3, в результате которой возникает дисфункция GABR, фенотипически проявляющаяся когнитивными расстройствами и судорожным синдромом. Shu-qun Shi и соавторы установили, что дефицит E6AP/UBE3A может приводить к нарушению циркадного ритма, который обусловлен увеличением представительства протеинов BMAL1 (brain-muscle-arnt-like protein-1)/ARNTL (aryl hydrocarbon receptor nuclear translocator-like 1) и BMAL2/ARNTL2. В настоящее время идентифицирован 21 ген, участвующий в поддержании циркадных ритмов организма человека (CLOCK, NPAS2, ARNTL1, ARNTL2, PER1, PER2, PER3, CRY1, CRY2, TIMELESS, NR1D1, RORA, RORB, RORC, CSNK1δ, CSNK1ε, GSK3β, DBP, BHLHB2, BHLHB3, PPARGC1A). С некоторыми генами данной группы ассоциированы генетические формы нарушения сна. Так, мутации гена PER2 сопряжены с развитием семейного синдрома смещения фазы сна (advanced sleep phase syndrome — ASPS). Циркадный ритм организма млекопитающих определен действием смены дня и ночи, света и тьмы. Основным морфологическим субстратом, который синхронизирует функциональную активность всех органов и систем со временем суток, является супрахиазматическое ядро гипоталамуса, воспринимающее сигналы, вызванные фоторецепцией сетчатки глаза. Возбуждение супрахиазматического ядра гипоталамуса приводит к активации факторов транскрипции CLOCK, BMAL1/ARNTL1. Данные факторы транскрипции, образуя единый комплекс, связываются со специфическими элементами ДНК E-box (5'-CACGTG-3 ') и E'-boxes (5'-CACGTT-3') промоторов генов-мишеней PER1, PER2 и CRY1, CRY2, продукты которых подавляют транскрипционную активность генов CLOCK, BMAL1/ARNTL1, формируя сеть отрицательных обратных связей. Наблюдаемое при AS снижение уровня деградации BMAL1/ARNTL1 обусловливает их накопление и увеличение длительности действия на целевые гены, что приводит к нарушению баланса времени бодрствования и сна за счет увеличения продолжительности времени бодрствования, которое сопровождается неэффективностью сна. Нарушение циркадного ритма функционирования клеток приводит к развитию метаболических расстройств. Большинство систем метаболизма человеческого организма подчиняется циркадному ритму. В многочисленных исследованиях показано, что изменение расписания рабочего времени сопровождается повышением заболеваемости диабетом, ожирением и частоты сердечно-сосудистых событий. Нарушение обмена глюкозы, липидов, накопление избыточного жира, расстройства термогенеза сопутствуют изменениям циркадного ритма. По всей вероятности, при AS нарушения молекулярных «часов» не ограничиваются только проявлениями расстройства сна.Также протеин E6AP/UBE3A действует как транскрипционный коактиватор рецептора стероидного гормона, участвует в регуляции клеточного цикла. Протеин E6AP/UBE3A регулирует трансактивность глюкокортикоидого рецептора (glucocorticoid receptor — GR) и его сигнального пути. У UBE3A-дефицитных мышей наблюдаются более высокий уровень кортикостерона в крови и недостаточная экспрессия нескольких GR-зависимых генов в некоторых областях головного мозга. Селективный дефицит GR сопровождается уменьшением количества парвальбумин-положительных ингибирующих интернейронов в гиппокампе и базолатеральной миндалине, что в конечном итоге способствует усилению стресса и тревожности.Схематически патогенез AS представлен на рис. 2.
|
Таким образом, морфологические или функциональные нарушения локуса q11-q13 копии материнской хромосомы 15 обусловливают потерю экспрессии гена UBE3A в нейронах головного мозга. Дефицит протеина UBE3A приводит к избыточному содержанию его целевых протеинов. Избыток протеинов ARC, эфексина-5 обусловливает нарушения цитоскелета дендритных шипов, что снижает синаптическую пластичность и проявляется когнитивными и поведенческими расстройствами, REST-ассоциированная дисфункция рецепторов гамма-аминомасляной кислоты обусловливает повышенную судорожную готовность, а избыточное содержание протеина BMAL1 приводит к нарушению сна и, возможно, трофики. http://www.mif-ua.com/archive/article/41548
#Семьям особенных детей нужна Ваша помощь#Благотворительный Фонд «Синдром Ангела»
|
|
|
|
|
|
|
|
 Помочьдетям.
Помочьдетям.