|
В поисках лечения синдрома Ангельмана.

|
В поисках лечения синдрома Ангельмана.Синдром Ангельмана - это неизлечимое заболевание нервной системы, вызванное нарушением функции материнской копии убиквитин-протеинлигазы E3A (UBE3A). Это редкое заболевание, которое встречается с частотой 1 на 12000-20000 рождений[1].Синдром был описан в 1965 году английским педиатром Гарри Ангельманом. Он сообщил о трех детях с похожими симптомами, которых он описал как похожих на марионеток детей с отрывистыми движениями и приступами смеха [2]В настоящее время отмечают следующие клинические особенности:100%: задержка развития; нарушение речи с отсутствием или малым количеством слов, рецептивные и невербальные навыки общения развиты лучше, чем вербальные; нарушения движения или баланса; частый смех/улыбки со счастливым выражением лица, легко возбудимые личности, гиперподвижное поведение, короткая продолжительность концентрации внимания;частые симптомы (>80%): задержанный, диспропорциональный рост окружности головы, обычно приводящий к микроцефалии в возрасте 2х лет; судорожные припадки, начало обычно в возрасте до 3х лет; ненормальная ЭЭГ, характерный паттерн с высокоамплитудными зубчатыми медленными волнами [3, 4]Ассоциированным симптомом является также нарушение сна и другие симптомы, которые встречаются в 20-80% случаев.
|

|
Рассмотрим генетику данного синдрома. У здоровых индивидов отцовская копия UBE3A является молчащей в нейронах из-за геномного импринтинга. В норме центр импринтинга PWS-IC метилирован на материнской хромосоме и деметилирован на отцовской хромосоме, при этом локус AS-IC управляет этим метилированием (рис.1) [5]. Рассмотрим сначала отцовскую хромосому. Отсутствие метилирования в центре импринтинга приводит к тому, что синтезируется ген SNHG14. Этот ген представляет собой сложный полицистронный транскрипт, состоящий из SNURF/SNRPN бицистронной матричной РНК, длинного некодирующего РНК транскрипта, который продуцирует набор малых ядерных РНК (SNORD107, SNORD64, SNORD109A, SNORD116, SNORD115 и SNORD109B) и UBE3A-AS (UBE3A-ATS) транскрипта. Интересно, делеция гена SNHG14, которая затрагивает SNORD109A и SNORD116 гены, вызывает другое нарушение развития нервной системы - синдром Прадера-Вилли. Антисмысловой транскрипт UBE3A-AS, расположенный на дистальном конце SNHG14 РНК путем неизвестного механизма блокирует экспрессию гена UBE3A с отцовской хромосомы [6]. Рассмотрим теперь материнскую хромосому. В ней участок PWS-IC метилирован, что приводит к подавлению синтеза гена SNHG14. В итоге не образуется антисмысловая РНК UBE3A-AS против UBE3A и ген UBE3A успешно экспрессируется (рис.1). |
|
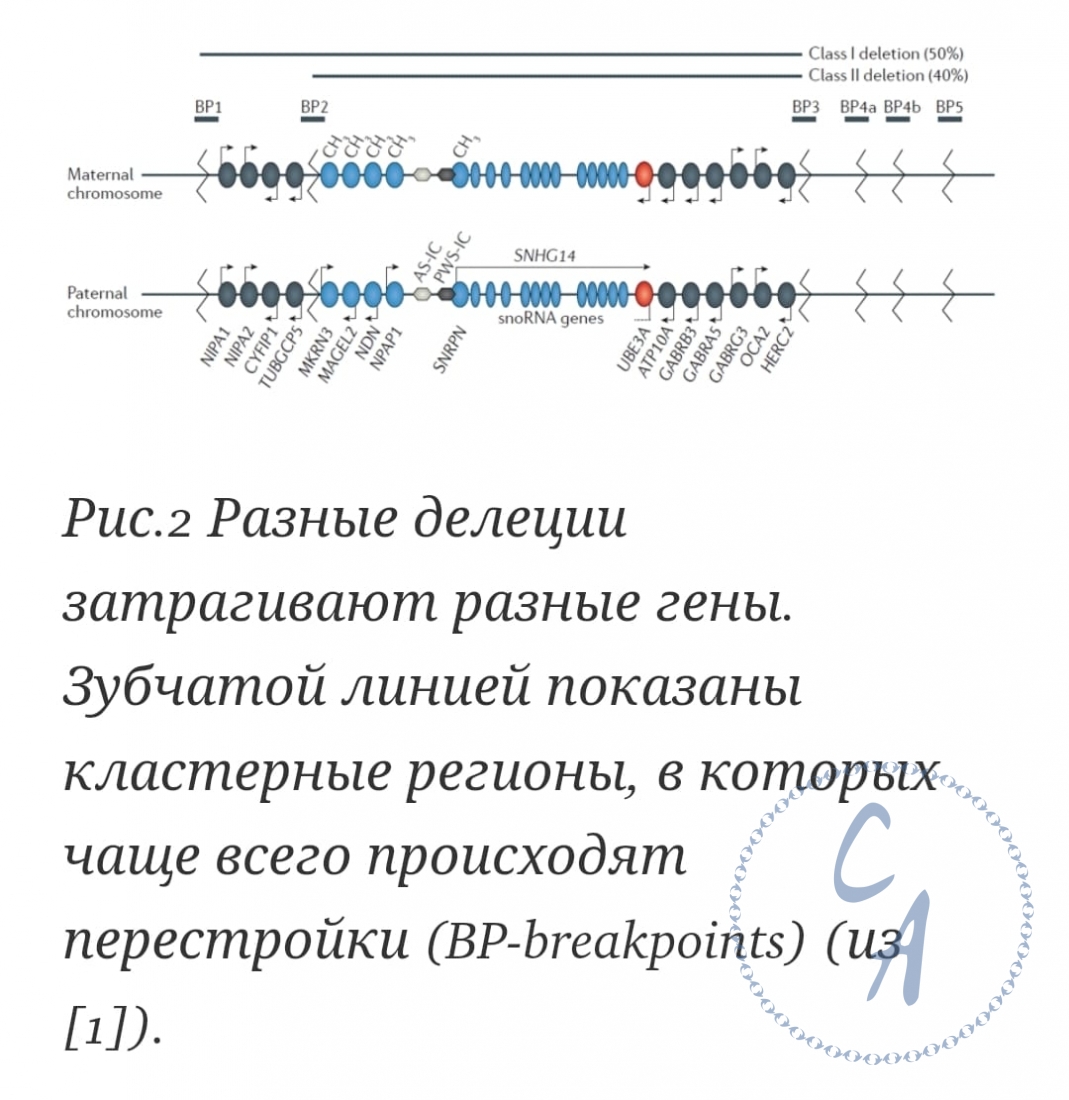
При синдроме Ангельмана экспрессия UBE3A может быть нарушена разными путями: делеция, затрагивающая материнскую копию UBE3A (около 70% случаев), патогенная мутация в гене UBE3A , дефект импринтинга, дисомия отцовской хромосомы 15 (без наличия материнской копии) - примерно по 10 % случаев каждый. Среди случаев с делецией, можно выделить два основных класса: Del1 - около 6Mб, около 16 генов и некодирующие регионы удалены, 40-50% делеций; и класс Del2 - около 5 Мб, 12 генов и некодирующие регионы удалены, 40-55% делеций. Атипичные делеции других размеров встречаются примерно в 5-10% случаев (рис.2) [1, 7]. Лица с делециями имеют более серьезные нарушения развития нервной системы, чем лица без делеций [7, 8]. Кроме описанных, при синдроме Ангельмана встречаются случаи мозаицизма, когда часть клеток нормальные, а часть - мутантные [1]. Убиквитинлигаза Ube3a - это фермент, который пришивает "метку смерти" убиквитин белкам, которые далее распознаются системой разрушения белков протеасомой и расщепляются внутри этого комплекса. Поэтому недостаток фермента Ube3a приводит к накоплению белков-мишеней, которые в норме должны быть разрушены. Существуют несколько стратегий для лечения синдрома Ангельмана, которые находятся на стадии доклинических и клинических исследований. Одна стратегия связана с восстановлением недостающего или нефункционального белка UBE3A в нейронах с помощью замены гена или замены фермента. Целью второго подхода является попытка "разбудить" отцовскую копию гена UBE3A. Третий подход связан с веществами, мишенью которых являются молекулярные пути и эффекторные белки, которые вовлечены в патофизиологию болезни Ангельмана [9]. Ниже перечисленные подходы во многом основаны на статье [9] с привлечением более новых данных. 1. Восстановление с помощью генной терапии. В одной из работ на мышах показано, что введение в гиппокамп аденоассоциированного вируса 9 типа (AAV9), содержащего копию гена UBE3A, восстанавливает ассоциативное обучение и улучшает память по сравнению с контролем, хотя электрофизиологические характеристики в гиппокампе были не полностью восстановлены. Стоит отметить, что ген эффективно не работал в мозжечке, и нарушения движений, которые, как предполагается, связаны с этой частью мозга, не были преодолены [10] Dr James Wilson на саммите FAST представил данные о том, что изоформа 1, но не изоформа 2 гена UBE3A, доставленная с помощью AAV, улучшала некоторые показатели активности у мышей с моделью синдрома Ангельмана, в том числе координацию движений [11]. Кроме того, вирус AAV-hu68-hUBE3A (изоформа 1) был введен макакам резус с целью оценки безопасности и уровня экспрессии в разных отделах мозга. Было показано широкое распространение вектора в мозге и отсутствие патологических процессов в спинном мозге у всех 250 исследованных обезьян [11]. Таким образом, доклинические исследования показали пригодность данного вида терапии для дальнейших клинических исследований. |

|
2. Клеточная терапия Клеточная терапия основана на способности стволовых гемопоэтических клеток (CD34+) дифференцироваться в разные типы клеток крови, включая иммунные. Хотя в норме микроглия в мозге имеет особое происхождение и не смешивается с макрофагами из циркулирующей крови, при воздействии некоторых химических веществ, например, бусульфана, как предполагается, происходит истощение пула микроглии в мозге и возникает возможность обмена с клетками крови [12]. Также важную роль играет механизм "кросс-коррекции", когда клетки могут обмениваться своим содержимым, и микроглия может передавать свое содержимое нейронам и глии (рис.4). Для клеточной терапии синдрома Ангельмана исследователи брали человеческие CD34+ клетки, трансдуцировали их лентивирусами с EGFP либо вектором, экспрессирующим модифицированную изоформу 1 гена UBE3A человека вместе с EGFP. Затем трансдуцированные гемопоэтические стволовые клетки инъецировали неонатальным и взрослым иммунодефицитным мышам с мутацией в гене UBE3A. Было показано восстановление функции движения и памяти у мышей с инъекцией клеток, трансдуцированных вирусом с геном UBE3A. Кроме того, улучшились показатели ЭЭГ в дельта-диапазоне. Также показана экпрессия гена UBE3A в мозге мутантных мышей [14, 15]. |

|
4. Активация "молчащей" отцовской копии гена UBE3A c помощью антисмысловых олигонуклеотидов. 4.1 Как было ранее сказано, отцовская копия гена UBE3A находится под действием антисмысловой UBE3A-AS последовательности. Данная последовательность является частью полицистронного гена SNHG14, в котором часть транскриптов является важной для работы нервной системы, поэтому репрессия целого гена SNHG14 может привести к негативным последствиям. Разработчики антисмысловых олигонуклеотидов GTX-102 выбирали участки на дистальном конце гена SNHG14 рядом с последовательностью UBE3A-AS для разрушения UBE3A-AS с помощью РНКазы Н (рис.3). Кроме того, выбранные последовательности являются консервативными в эволюции, поэтому можно было протестировать их на макаках. На обезьянах показано, что отцовская копия UBE3A экспрессируется в моторной, фронтальной коре и спинном мозге нескольких животных, при этом полная биаллельная экспрессия достигнута в спинном мозге шести животных после интратекальной инъекции [6]. В настоящее время препарат GTX-102 находится на стадии клинических испытаний, и первые результаты являются обнадеживающими [16, 17]. Недостатком антисмысловых олигонуклеотидов является необходимость многократного введения лекарства в спинномозговую жидкость. 4.2. ION582 - представитель 2'-О-метоксиэтил антисмысловых олигонуклеотидов, которые сконструированы для большего подавления мишеней и лучшей переносимости. Действует по РНКаза Н механизму [18]. Авторы исследования начали со скрининга 3000 антисмысловых последовательностей в разных моделях для поиска наиболее эффективной молекулы. В настоящее время ION582 находится в активной фазе поиска пациентов для проведения 1 фазы клинических исследований [19]. 4.3. Ругонерсен - антисмысловой нуклеотид от компании Roche, который химически модифицирован с LNA (locked nucleic acid). В настоящий момент находится на 1/2 фазе клинических исследований. На обезьянах показано, что введение ругонерсена вызывает увеличение мРНК и белка гена UBE3A и падение антисмысловой последовательности. Проведенные PET исследования показали, что ругонерсен хорошо распространятеся в спинном и головном мозге [20, 21] 5. Нокдаун антисмысловой последовательности с помощью геномного редактирования с помощью CRISPR/Cas9 [22-24], чтобы постоянно инактивировать UBE3A-ATS. Гидовая РНК расположена проксимально от антисмысловой последовательности. На мышиной модели синдрома Ангельмана показано частичное восстановление экспрессии Ube3a и частичное восстановление поведенческих функций [9]. |
6. Нокдаун антисмысловой последовательности с помощью микроРНК [25]. Доставка генетического материала для синтеза микроРНК осуществлялась с помощью внутримозговой инъекции в мозг мышей адено-ассоциированного вируса. Несмотря на высокий уровень микроРНК, восстановление синтеза белка не очень высокое по данным иммуногистохимии. Помимо описанных методов, для индукции экспрессии отцовской копии UBE3A гена предлагается использовать искусственные транскрипционные факторы и ингибиторы топоизомераз [9]. Кроме воздействия на ген UBE3A, существуют подходы к терапии на основе нижележащих путей или симптомов заболевания [9]. 1. Циклический глицил-пролин (NNZ-2591). Природный метаболит IGF-1, который регулирует его биодоступность. NNZ-2591 - синтетический аналог цГП, который имеет более длительный период полураспада и улучшенную биодоступность. 6-недельное лечение мышей с моделью синдрома Ангельмана привело к значительному улучшению моторного и когнитивного дефицита и снизило количество судорожных событий. Фаза 1 клинических исследований показала безопасность препарата у здоровых волонтеров. Начата 2 фаза клинических исследований у пациентов с синдромом Ангельмана [26, 27]. 2. На мышиной модели синдрома Ангельмана показан эффект в поведенческих экспериментах лигандов IGF2-рецептора [28]. Однако позже другой командой было показано, что Igf2 влияет на электрофизиологические характеристики только в остром эксперименте, однако длительного действия или эффекта на поведение обнаружено не было в мышиной и крысиной моделях синдрома Ангельмана [29]. Возможно, требуется более подходящая дозировка, либо Igf2 не подходит для терапии симптомов синдрома Ангельмана. Стоит отметить, что исследования Igf2 на крысах нельзя напрямую переносить на человека, поскольку у человека экспрессия эндогенного Igf2 отличается от таковой у крыс [30]. 3. Помимо UBE3A, при делециях нарушается функция и других генов, например, ГАМК-рецепторов. Предполагается, что нарушение ГАМК-рецепторов ответственно за более частые случаи эпилепсии при делециях участка хромосомы 15 по сравнению с другими генотипами синдрома Ангельмана без делеций. Алогабат - позитивный аллостерический модулятор альфа5-ГАМКа-рецепторов, который усиливает передачу сигнала во время связывания ГАМК с рецептором, что должно усиливать торможение в нервной системе и уменьшать вероятность эпилептических эпизодов. В настоящее время готовится 2я фаза клинических исследований для изучения влияния алогабата на показатели ЭЭГ у детей и подростков с синдромом Ангельмана [31, 32]. 4. Также исследуется препарат SAGE-324, позитивный аллостерический модулятор ГАМК рецепторов. Препарат находится на 2 фазе кличнических испытаний для лечения тремора [9, 33]. 5. Ингибитор фосфатазы PP2A - LB-100. Активатор PP2A является субстратом UBE3A, и поэтому пациенты с синдромом Ангельмана имеют ненормально высокий уровень активности PP2A. Генетическое снижение уровня активатора или фармакологическое ингибирование PP2A восстанавливало морфологию дендритных шипиков в мышиной модели синдрома Ангельмана. Ингибитор LB-100 улучшил синаптическую трансмиссию в первичной моторной коре мышей с синдромом Ангельмана. Кроме того, инъекция LB-100 этим мышам приводила к значимому улучшению мышечной силы, двигательной координации и обучения через 14 дней [34]. Для LB-100 показана безопасность в фазе 1 клинических испытаний для лечения солидных опухолей [35]. Эта молекула сейчас проходит тестирование на способность преодолевать гематоэнцефалический барьер у пациентов с опухолями мозга [36]. 6. Другой мишенью Ube3a являются калиевые SK2 каналы. Убиквитинлигаза Ube3a вовлечена в удаление этого канала из синаптических областей внутрь клетки [37, 38] . Это приводит к тому, что при нарушении работы убиквитинлигазы Ube3a калиевые каналы накапливаются в синаптических областях. Блокада калиевых каналов предотвращала нарушение долговременной потенциации, наблюдаемое в мышиной модели синдрома Ангельмана. Кроме того, в мышиной модели синдрома Ангельмана наблюдалась усиленная долговременная депрессия, и блокатор калиевых каналов апамин также возвращал значения к здоровому контролю. Также блокатор калиевых каналов апамин улучшил обучение мышей с синдромом Ангельмана в модели условно-рефлекторного страха. 7. NSI-189 phosphate. NSI-189 фосфат - это нейропротективный агент, для которого была показана способность стимулировать нейрогенез как in vitro в культуре, так и in vivo в гиппокампе грызунов. В фазе 2 клинических исследований для терапии большого депрессивного расстройства [39] NSI-189 показал антидепрессантный и прокогнитивный эффекты. В доклинических исследованиях синдрома Ангельмана на срезах мозга мышей показано улучшение в одном из видов долговременной потенциации в гиппокампе. Кроме того, лечение мышей этим препаратом в течение 16 дней показало улучшенное обучение и память в модели условно-рефлекторного страха. Лечение в течение 5 дней показало улучшенную двигательную функцию. После лечения в течение нескольких дней эффекты сохранялись в течение более трех недель, хотя период полужизни вещества составляет у мышей порядка 2 часов. Предполагается, что эффекты связаны с изменением в транскрипции генов [40]. 8. Кетоновые эфиры. Увеличение уровня кетонов путем ограничения потребления углеводов менее 10 грамм в день успешно использовалось для лечения эпилепсии у одного пациента с синдромом Ангельмана [41]. Ограничение углеводов с низким гликемическим индексом до 40-60 грамм в день у пациентов с синдромом Ангельмана показало хорошие результаты в предотвращении эпилептических припадков [42]. В рекомендациях по уходу за пациентами с синдромом Ангельмана, сделанных учеными и педиатрами разных стран, кетоновая диета или диета с низким гликемическим индексом показана для разных возрастных групп [43]. Альтернативой кетогенной диете является употребление кетоновых эфиров. В доклинических исследованиях употребление кетоновых эфиров снизило индуцированную эпилептическую активность, улучшило поведение и гиппокампальную синаптическую пластичность у мышей с моделью синдрома Ангельмана [44]. Однако, добавка кетонового эфира бета-гидроксибутирата на второй фазе клинических исследований показала скромные результаты. Так, несмотря на множество измеряемых параметров, достоверные различия получены только для малого количества из них, что объясняется авторами возможно малой выборкой, поскольку основной целью было показать безопасность препарата, что они и сделали. Авторы показали улучшение в мелкой моторике, а также некоторое улучшение в задаче на слуховую память, сделанную с помощью измерения фронтального потенциала. Эффект на эпилептические припадки не анализировался, поскольку только у четырех пациентов в базовом периоде наблюдались судорожные события [45]. Таким образом, в настоящее время существует много подходов к возможному лечению синдрома Ангельмана как со стороны генной терапии, так и со стороны малых молекул, однако все они находятся на той или иной стадии докличнических испытаний или клинических исследований. Большую роль в развитии новых методов лечения играет организация FAST (Foundation for Angelman Syndrome Therapeutics; https://cureangelman.org), которая организована родителями детей с синдромом Ангельмана вместе с профессионалами для поиска новых средств лечения синдрома. Кроме того, исследованиями синдрома Ангельмана занимается организация Angelman Syndrome Foundation (angelman.org). В нашей стране также занимаются исследованиями синдрома Ангельмана [4, 46], однако исследований новых видов терапии этого заболевания в России в процессе подготовки статьи найдено не было. Наталья Баль, https://t.me/neuronsandsynapsesJanuary 20, 2023
#Благотворительный Фонд помощи людям с синдромом Ангельмана «Синдром Ангела» #помочь особенным детям
|
|
|
|
|
|
|
|
 Помочьдетям.
Помочьдетям.















